研究背景
尖晶石LiNi0.5Mn1.5O4(LNMO)正极材料具有较高的氧化还原电位和高能量密度(~650Whkg−1)。然而,LNMO的高工作电压导致其与电解质发生副反应,导致正极界面的退化,导致循环时容量下降。碳酸盐电解质在4.4 V 电压下在正极表面形成厚且不稳定的正极-电解质间相(CEI),较厚的CEI层具有较高的界面电阻,从而损害了正极的循环稳定性。因此,LMNO正极在高电压下的循环稳定性一直被认为是一种不可避免的问题。另一种有害的降解机制涉及六氟磷酸锂(LiPF6),在高电压下,LiPF6可以与微量的水反应生成氟化氢(HF),从而引发过渡金属(TM)离子从正极表面的溶解和迁移。当裸露的LNMO表面暴露在电解质中时,TM离子的溶解会加速。溶解的TM离子沉淀在负极/正极,以金属粒子形式的表面进一步催化两个界面上副反应。迄今为止,已经提出了各种抑制电解质副反应的策略,如使用电解质添加剂、自由基清除剂、和界面保护层。然而,完全覆盖活性粒子是不可行的,即使可以实现完全覆盖,也会提高界面阻力。自由基清除剂不能完全消除HF分子。另一方面,粘结剂具有保护活性电极层和集流体之间的粘附力,因此,我们假设粘合剂与电解液可能对正极界面的形成和性能产生重大影响,而正极界面与电池的关键电化学性能指标密切相关。
虽然聚(偏氟乙烯)(PVDF)是最广泛使用的粘合剂层状金属氧化物正极,但并不适用于LNMO正极,因为其不能均匀覆盖活性物质,可能无法解决上述界面问题。作为PVDF的替代品,生物聚合物和合成聚合物利用它们的羟基或负电荷官能团分别参与与LNMO表面的氢键和离子偶极相互作用。这些相互作用排列均匀地覆盖LNMO表面,从而防止副反应发生。然而,这些粘合剂的均匀覆盖,反过来又增加了界面电阻,因为从结构的角度来看,它们对锂离子的电导率造成不利影响。粘合剂有一个关键的功能是影响锂离子扩散到CEI层,氧化弱官能团粘结剂在高压下可以转换成某些部分CEI层。考虑到这一原理,可以选择粘合剂的官能团,使分解后剩余的部分形成CEI配合物/粘合物,以促进锂离子在CEI中的扩散。因此,粘合剂可以表现为“牺牲性的”。虽然电解质添加剂也可以设计出类似的目的,但如果其最低未被占用的分子轨道水平低于负极的工作电位,则干扰负极界面。显然,粘合剂的主要作用必须通过利用其他官能团来保持其粘附性能,强调多功能性作为LNMO正极粘合剂的关键设计原则。
成果简介
近日,首尔国立大学/韩国能源研究所等合作者为了解决LNMO高电压循环下容易发生界面降解副反应,从而阻碍长期和高倍率循环稳定性等问题。在此,作者通过掺入牺牲性粘合剂,即 λ-卡拉胶 (CRN)(一种硫酸化多糖),克服了LNMO长期循环性较差的问题。这种粘合剂不仅通过氢键和离子偶极相互作用均匀地覆盖LNMO表面,而且提供了含有LiSOxF的离子导电正极-电解质界面层。利用这两个特性,CRN基电极的循环性能和倍率性能远远优于基于传统的聚(偏二氟乙烯)和海藻酸钠粘合物。本研究引入了一种新的概念,即“牺牲”粘合剂,用于锂离子电池电极制造,使其具有优越的电化学性能。该工作以“Carrageenan as a sacrificial binder for 5 V LiNi0.5Mn1.5O4 cathodes in lithium-ion batteries”为题发表在Advanced Materials上。
研究亮点
(1) 通过使用硫酸盐多糖粘合剂,即CRN,克服了LNMO电池在高压循环条件下稳定性较差的问题;
(2) 粘合剂形成CEI/粘合物,通过高压条件下硫酸盐基的不可逆氧化分解对锂离子具有高导电性;
(3) CRN的亲水性使其可行的覆盖LNMO粒子均匀,但硫酸基的氧化分解导致CEI/粘合剂复杂层包含LiSOxF,促进锂离子传导;
(4) 本研究是首次在粘合剂设计中演示“电化学牺牲”概念来控制CEI层。
图文导读
CRN在其主主链上同时有硫酸基和羟基(图1a),使其能够同时作为牺牲剂和粘合剂。这两种功能在5V 高压下同时解决LNMO正极两个挑战性问题:基于正极界面的强粘附性和高锂离子传导,具有良好的活性粒子覆盖范围。为了证明CRN这两个官能团的作用,我们选择了海藻酸钠(ALG,图1b)和PVDF(图1c)作为对照结合剂。ALG在结构上与CRN高度相似,只是ALG没有硫酸盐基团。ALG长期以来一直被用作正极和负极的水性粘结剂。利用透射电子显微镜(TEM)研究了原始状态下LNMO表面粘合剂的覆盖情况,CRN和ALG粘结剂沿LNMO表面的覆盖范围较薄且均匀(图1d,e),这归因于它们的亲水官能团。相比之下,PVDF的分布并不均匀,因为它在LNMO表面局部聚集(图1f)。这是因为PVDF具有弱范德华相互作用,没有引起与活性材料的粘附。
为了证明CRN中的硫酸盐官能团确实被电化学分解,在第一个和第三个形成周期中,在半电池中进行了电化学浮充试验(图1g-j)。在这些浮充测试中,在每个电压水平上施加恒定电位10小时,同时监测特定的电流。第一个形成周期的充电过程的特征是ALG基和PVDF基电极都有连续的无峰曲线(图1g)。然而,对于基于CRN的电极,在4.8 V时观察到一个肩峰(图1g中的箭头),这在图1h的放大图中更明显。仅在CRN基电极的曲线上出现这个肩峰是由于其硫酸盐官能团的氧化分解,而不是与电解质分解相关。在第三个形成循环中,所有电极的电流曲线形状相似(图1i,j),这表明在初始充电过程中CRN的氧化是不可逆的,之后电极稳定。这一结果与电解质添加剂的报道一致;从CRN中提取的硫酸衍生物在初始充电过程中在LNMO表面形成,一旦在早期循环中建立了电子绝缘界面层,就不会被进一步氧化。这些一系列的结果表明,CEI/粘合剂配合物的稳定性在第一次循环中形成,粘合剂在该配合物的稳定性中起着至关重要的作用。
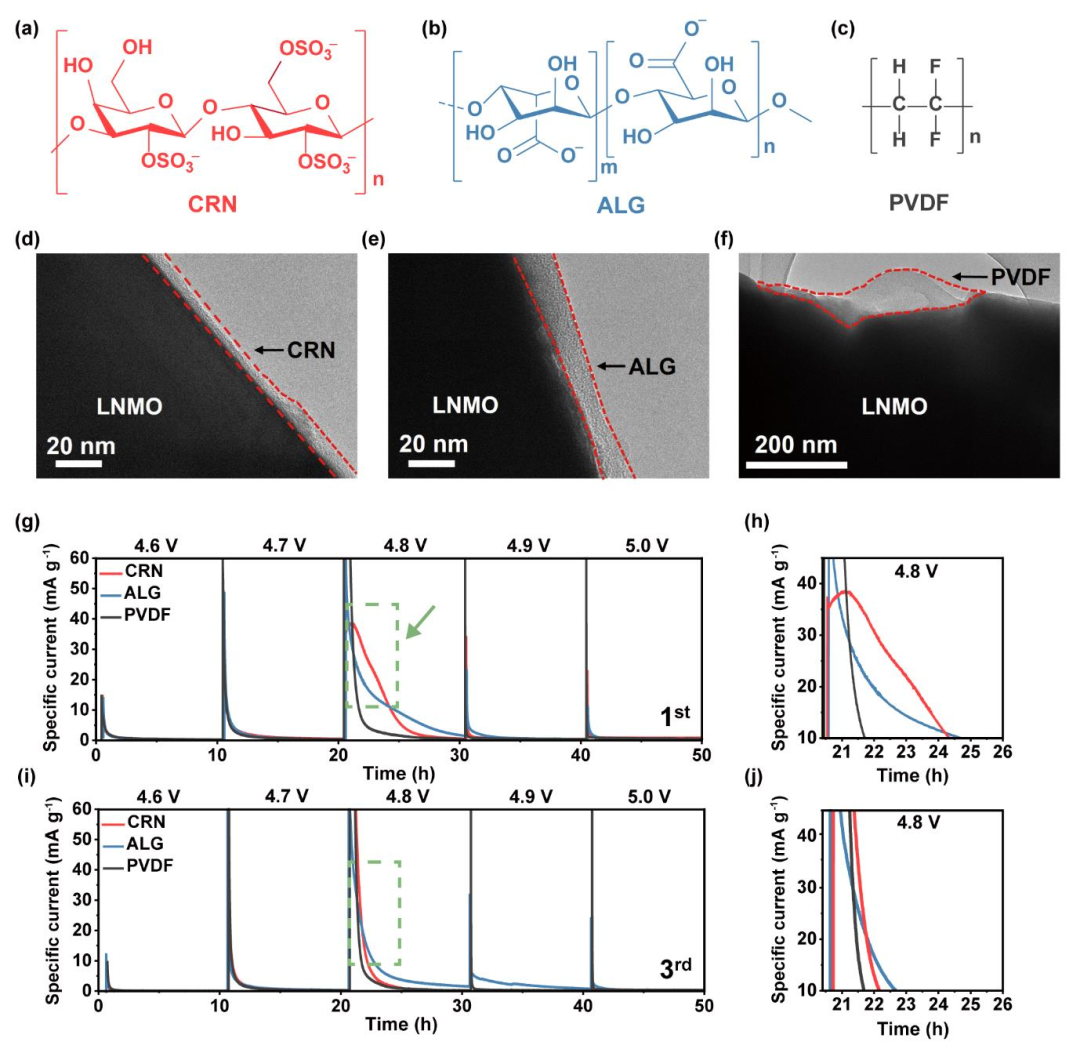
图1.CRN、ALG和PVDF聚合物粘合剂的理化和电化学性质。(a) CRN、(b) ALG和(c) PVDF的化学结构。在原始状态下,LNMO表面上的(d) CRN、(e) ALG和(f) PVDF的透射电镜图像。红色虚线表示粘合剂所覆盖的表面区域。在(g和h)第一次和(i和j)第三次充电时,CRN、ALG和PVDF基电极的电化学浮动试验周期(h和j)分别由(g和i)中绿色虚线划分的区域的扩大
图2a显示了三组电池在经过活化3圈后在1C(1C = 147 mA g−1)和室温下在3.5-5.0V电位范围下的循环性能。基于CRN的电池在循环性和比容量方面都比基于ALG和PVDF的电池表现出优越的性能。基于CRN-、ALG-和PVDF-的电池分别提供了110.7、96.1和112.0mAhg−1,在900次循环后保留了100.0%、98.6%和75.4%的原始容量。三组电池的平均CEs分别为99.6%、99.6%和99.4%。多糖粘合剂对LNMO表面的均匀覆盖可以缓解表面降解,从而更有效地维持循环。硫酸基分解引起的CEI的离子导电特性使其具有更高的比容量。粘合剂也会显著影响倍率性能。在0.1C到5C之间,基于CRN电池的放电容量保持得要高得多(图2b),明显利用了其CEI/粘合剂配合物高的锂离子电导率。例如,在5C时,CRN、ALG和PVDF基细胞的比容量分别为81.3、59.7和36.3 mAh g−1。通过电化学阻抗谱(EIS)对原始状态和100个循环状态的分析,阐明了三组电池之间不同的倍率性能(图2c,d)。CEI电阻(RCEI)和电荷转移电阻(RCT)与LNMO表面的界面电阻密切相关。在原始状态下(图2c),CRN基电极比PVDF基电极表现出更高的界面电阻(CRN基和ALG基电极的界面电阻分别为305.0和343.1Ω),而PVDF基电极为139.5 Ω)。经过100个周期后(图2d),基于CRN的细胞的RCEI和RCT(4.5和88.9 Ω)分别低于基于ALG电极(分别为7.2和155.1Ω)和基于PVDF的细胞(分别为64.2和252.9Ω)。这一结果再次反映了CRN中硫酸盐基团形成的CEI/粘合剂配合物的锂离子电导率较高。在100次循环中,PVDF电极的RCEI和RCT均显著增加,这反映了PVDF覆盖范围有限导致界面不稳。此外,在0.05到1 mV-1的不同扫描速率下的CV被用来研究锂离子在LNMO表面的传输。锂离子扩散系数(DLi+)可以从ip和v1/2之间的关系中提取出来,用如下式表示:
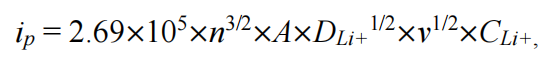
ip是峰值电流,n是电子反应的数量(n=1),A为电极的表面积,v是扫描速率,CLi+为锂离子的体积浓度(0.02378 mol cm–3)。利用ip与v1/2图的斜率,计算出基于CRN-、ALG-和PVDF-电极的DLi+值分别为2.84×10-10、1.81×10-10、8.98×10-11 cm2s-1(图2e)。这些结果与倍率性能和EIS结果所表现出的趋势一致,并归因于锂离子通过CRN基电极的锂离子导电CEI/粘合物的高效迁移。
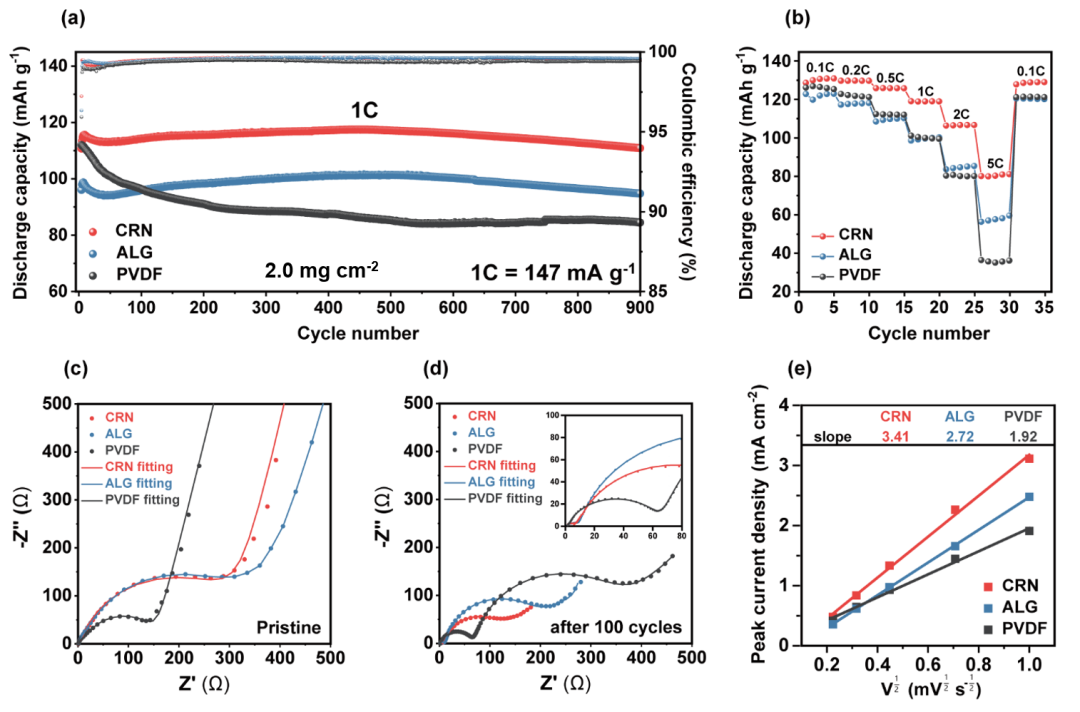
图2. CRN-、ALG-和PVDF-基电极的电化学性能。(a)在3.5-5.0V(14.7 mA−1)下进行900个循环的放电容量和CEs。半电池0.1C(14.7 mA g−1)下形成三次循环。(b)在不同倍率下的倍率性能。奈奎斯特图在原始状态的(c)和100个循环后拟合。(e)峰值电流(ip)与CV数据中扫描速率(v1/2)的平方根之间的关系。
图3a-c分别显示了CRN-、ALG-和PVDF-基电极在三个不同循环阶段的F 1s XPS谱:原始阶段、三个形成循环后和100个循环后。在原始状态下,CRN-和ALG-基电极没有观察到信号;然而,在PVDF基电极中观察到一个属于PVDF的强C-F键的峰值。经过三个形成周期后,在基于CRN-和ALG-的电极上观察到与氟化锂相关的峰(图3a,b),这是由于PF6-阴离子的分解。CRN-电极在687.98 eV处出现了一个峰值,分配给LiSOxF。通过与商业LiSO3F的比较,证实了该峰的存在。相比之下,这些无机物的峰在PVDF基电极的光谱上没有明显的特征(图3c)。经过100个循环后,CRN-和ALG-基电极中的氟化锂下降(图3a,b)。CRN-基电极中的LiSOxF也降低,相比之下,PVDF-基的电极在相同的循环期间,氟化锂和LixPOyFz均表现出更大的增加(图3c)。考虑到XPS的探测深度约为5nm,预计在循环的进行,CRN-和ALG-基电极早期形成的氟化锂逐渐被其他CEI覆盖。与此相反,在PVDF基电极的情况下,氟化锂和LixPOyFz都随着循环的增加而增加。
在其S 2p XPS结果中进一步捕获了CRN基电极的CEI特性。在原始状态下,观察到与O-SO3键相关的双峰峰(171.17和169.97eV)(图3d),反映了裸CRN中的硫酸盐基团。3个循环后,S 2p的LiSOxF峰值和O 1s XPS谱下降,类似于多糖电极的氟化锂峰,表明这些组分在LNMO表面形成,但在以后的周期中几乎没有形成。此外,即使在循环后,CRN基电极中的硫酸盐(O-SO3)峰仍然被检测到,这意味着CRN中并非所有的硫酸盐都被消耗掉以形成LiSOxF。二次离子质谱飞行时间(ToF-SIMS)也检测到LiSOxF的存在(图3e,f)。对离子碎片的质谱分析显示,LiSO3F-(m/z=105.97)和LiSO2F-(m/z=89.98)以负离子形式存在,作为LiSOxF的特征。电化学浮动测试结果、O 1s、F 1s和S 2p XPS结果以及ToF-SIMS结果一致支持我们的观点,即CRN电极中的CEI层包含高压下CRN硫酸官能团分解的LiSOxF。
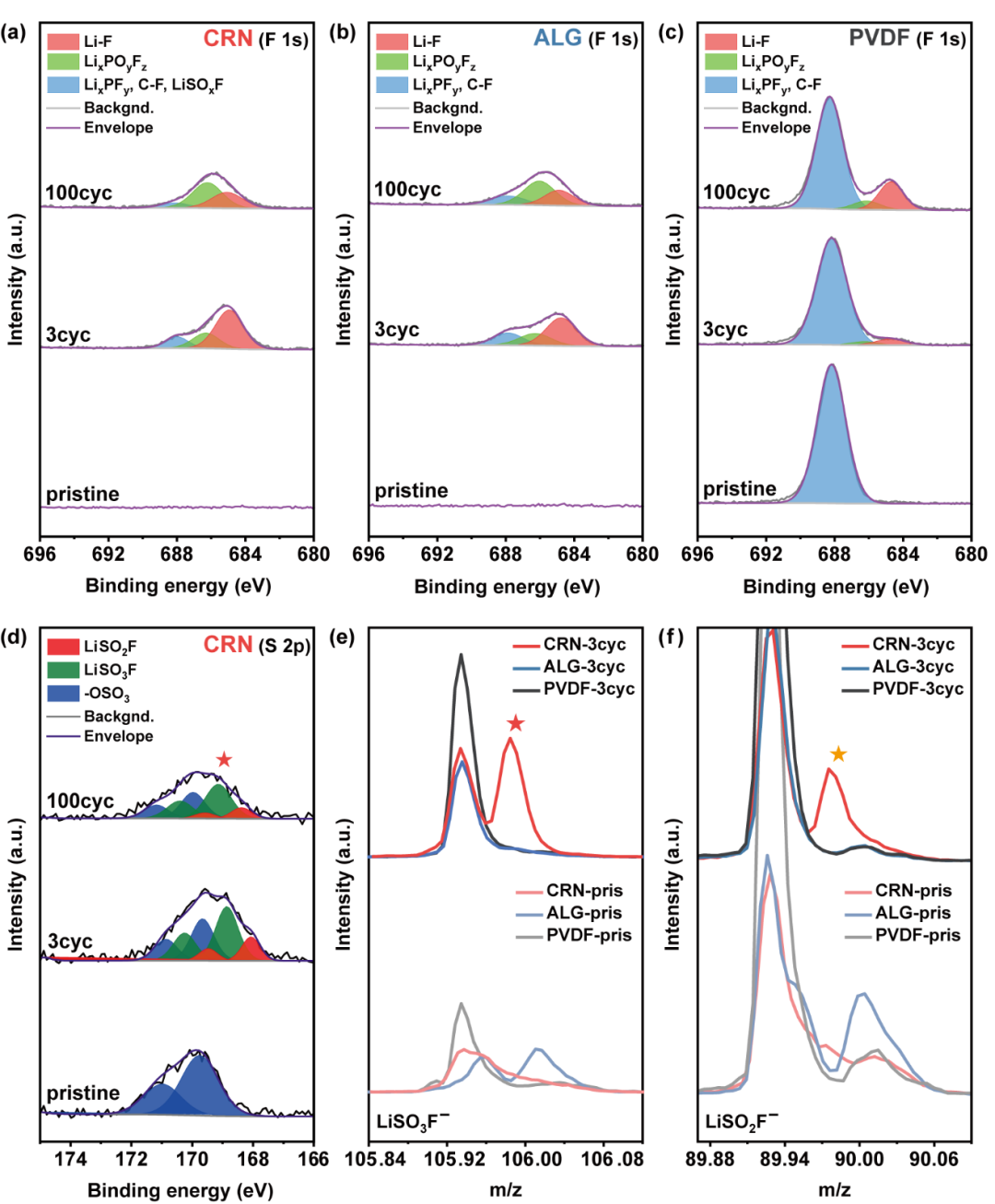
图3.CRN-、ALG-和PVDF基电极中CEI组分的识别。本研究中(a) CRN-、(b) ALG-和(c)PVDF基电极的F 1s XPS结果在原始状态,在0.1C下三次循环,在1C下100次循环。(d) S 2p XPS结果在原始状态下,在0.1C下三次循环,在1C下100次循环。(e) LiSO3F−和(f) LiSO2F−二次离子片段在原始状态下的ToF-SIMS质谱。星号标记的峰代表LiSO3F−(红色)和LiSO2F−(橙色)离子片段(Li=7.016,S=31.972,O=15.994和F=18.998)的m/z特征值。
追踪其他阴离子LiSO2F- 和LiF-(m/z=26.00)碎片,作为溅射时间的函数(图4a)。与S 2p XPS结果一致,LiSO2F- 阴离子片段的强度随着溅射开始开始增加,40秒后阴离子片段的强度逐渐下降,表明其更集中地存在于CEI内部。仔细观察,在CRN基电极中,溅射40秒后LiSO2F-阴离子片段的强度高于溅射前(图4b)。这与基于ALG-和PVDF的电极不同(图4c,d),它们在相同的溅射时间内的峰值强度几乎没有差异。与CEI/粘合物的氟化锂成分相关的LiF-阴离子片段的深度剖面也显示了类似的异常。与基于CRN和ALG的电极不同,其强度在初始40秒内增加,然后逐渐下降。基于PVDF的电极中的LiF-的强度从一开始就单调下降。从三个电极在0和40秒时的锂离子片段的强度曲线上可以明显地看出这些趋势(图4e-g)。同样,该阴离子片段的峰值强度与CEI/粘合物内部存在的浓缩氟化锂有关。ToF-SIMS深度剖面数据描绘了一个关于CRN基电极的CEI层的一致图像。也就是说,CEI层的特征是在该层的内部形成具有有益的LiF和LiSOxF,这在保护电极免受有害的副反应方面起着重要的作用。根据目前所得到的分析结果,三个电极的CEI层的组成和结构如图5所示。
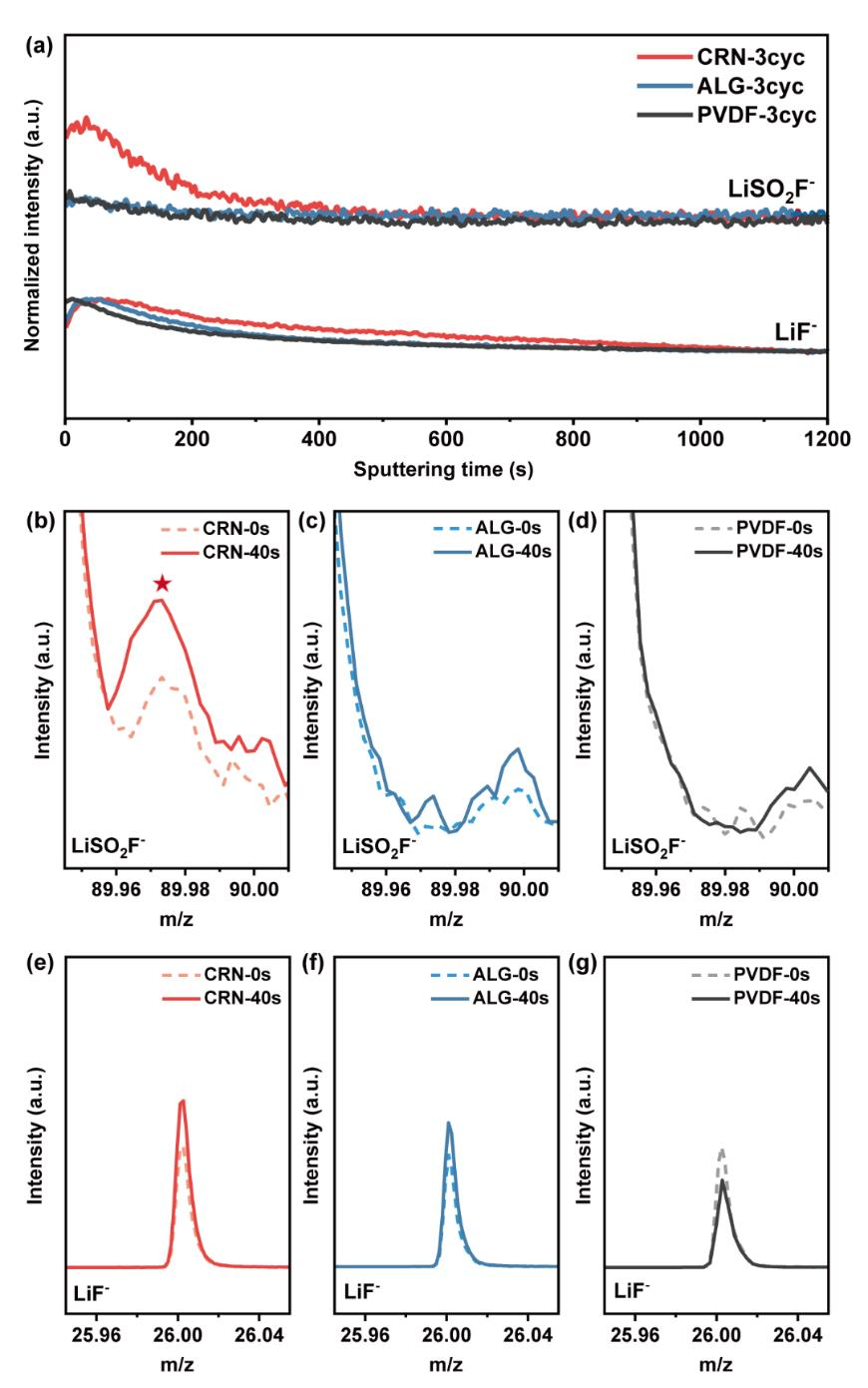
图4. 无机CEI组分在CRN-、ALG-和PVDF-基电极中的不同位置。在0.1C下,三个粘结剂电极的LiSO2F−和LiF−二次离子片段的(a) ToF-SIMS深度分布。在0和40秒时,(b) CRN-、(c) ALG-和(d)PVDF电极中LiSO2F−二次离子片段的ToF-SIMS谱。(e) CRN-、(f) ALG-和(g)PVDF基电极中LiF−二次离子片段的ToF-SIMS谱。

图5.PVDF-、ALG-和CRN基电极的粘合剂覆盖范围和CEI形成的示意图
在锂离子扩散方面,CRN基电极CEI层内部的LiSOxF/LiF的组合组成比ALG基电极CEI层内部的LiF更有利。这与CRN基电极优越的放电容量和倍率性能相一致。这些比较结果使我们分别考虑LiSOxF和LiF的锂离子和反阴离子SOxF−和F−之间的静电学。因为静电力会影响CEI层中锂离子迁移率。为此,通过密度泛函理论(DFT)计算,计算了锂离子与反阴离子的结合能(图6a)。计算结果表明,CRN中的硫酸锂衍生物(LiSO3F和LiSO2F)的结合能分别为-6.27和-6.59eV,均低于氟化锂(-8.11eV),说明Li离子相互作用与LiSOxF的阴离子比LiF强。此外,利用固态7Li核磁共振波谱(7LiNMR)对商用LiSO3F和氟化锂进行了分析,实验验证了上述计算结果(图6b)。根据7Li-NMR分析,LiSO3F的化学位移(-1.44ppm)低于氟化锂(-0.85ppm),说明LiSO3F中Li原子附近较高的电子密度进一步屏蔽了Li原子的核不受外场的影响。这一结果与上述DFT计算结果相关联,即LiSO3F的阴离子与锂离子的结合较弱,从而使锂离子可以更自由地扩散。
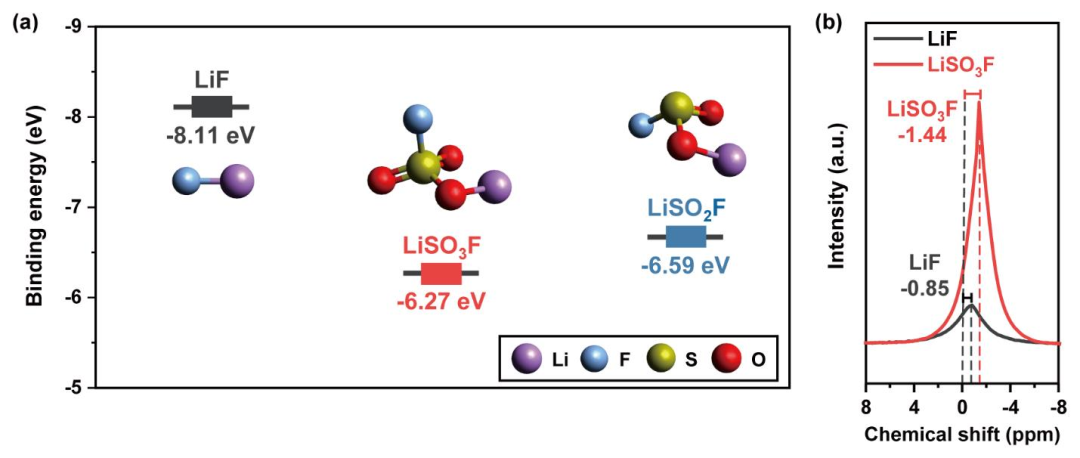
图6. LiF和LiSOxF(x = 2,3)CEI组分的锂离子结合亲和力。利用GAUSSIAN16软件包,通过DFT计算得到LiF和LiSOxF(x = 2,3)的(a) Li离子结合能。氟化锂和LiSO3F的(b) 7Li MAS核磁共振谱。
总结与展望
在这篇文章中,作者通过使用硫酸盐多糖粘合剂,即CRN,克服了LNMO正极在高电压下表面降解导致循环稳定性较差的挑战。这种粘合剂形成CEI/粘合物,通过高压条件下硫酸盐基的不可逆氧化分解对锂离子具有高导电性。CRN的亲水性使其均匀覆盖LNMO粒子,硫酸盐基的氧化分解导致CEI/粘合剂复杂层包含LiSOxF,促进锂离子传导,解决LNMO正极在高倍率和长循环条件下的问题。本研究是首次在粘合剂设计中演示“电化学牺牲”概念来控制CEI层。基于同样的原理,可以考虑各种官能团来靶向改性锂离子电池正极,解决界面不稳定性带来的不利影响,使这些电极具有优越的电化学性能。
审核编辑:刘清